
第一作者:贾欠欠
通讯作者:黄理志
通讯单位:js6666zs金沙
图文摘要
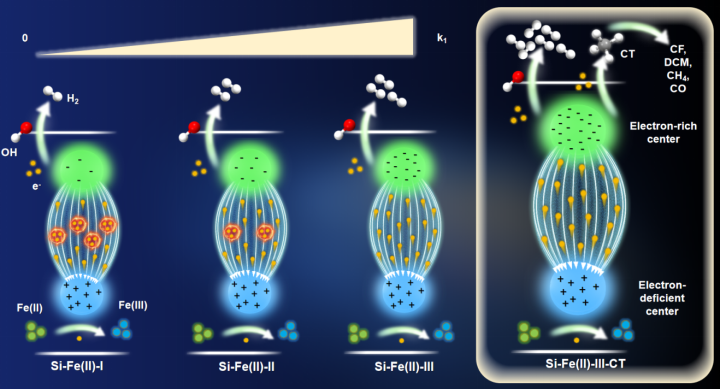
成果简介
近日,js6666zs金沙市政系黄理志团队在Water Research上发表了题为“Reductive dehalogenation in groundwater by Si-Fe(II) co-precipitates enhanced by internal electric field and low vacancy concentrations”的论文。该项工作研究了不同羟基化顺序合成的Si-Fe(II)共沉淀物对有机污染物的还原脱氯机制研究。在有氧条件下Fe(III)和Si之间的相互作用已被广泛研究,但在缺氧和还原条件下Fe(II)和Si之间的相互作用研究很少。在天然地下水环境中,Si和Fe(II)的相互作用是多样的。然而,这种相互作用对Si-Fe(II)共沉淀物的结构和相组成的影响在很大程度上被忽略了。因此,对Si-Fe(II)共沉淀物的结构信息、反应性及其对地下水污染物转化的影响研究较少。这项工作加深了我们对自然地下环境中丰富的Si-Fe(II)相的稳定性、相变、还原反应活性和应用的理解。
引言
铁(Fe)是地壳中含量第四高的元素,几乎存在于所有的水环境中。而硅(Si)占地壳的27.7%,使Si成为仅次于第二丰富的氧元素。因此,硅化合物通常存在于土壤和地下水环境中。溶解硅酸盐也是天然水中的重要成分。Fe和Si之间的相互作用在许多地球化学循环中起着重要作用,尽管Fe(II)和Si之间的相互作用已在有氧条件下进行了广泛研究,但在缺氧和还原条件下Fe(II)和Si的相互作用很少被研究。在天然地下水环境中,Si和Fe(II)的相互作用是多样的。然而,这种相互作用对Si-Fe(II)共沉淀的结构和相组成的影响在很大程度上被忽略了。虽然已知Si-Fe(II)共沉淀相是层状硅酸盐和SiO2,但在原子尺度上尚未清楚地了解Si-Fe(II)共同沉淀的结构信息。因此,很少研究Si-Fe(II)共沉淀的反应性及其对地下水污染物转化的影响。Fe和Si物种的分布强烈取决于溶液pH。羟基化序列可以改变Fe(II)的配位环境,从而导致Fe(II)八面体结构的不同变形程度。这使得Fe(II)的给电子能力被改变。因此,本文研究了羟基化顺序对Si-Fe(II)共沉淀反应性的影响。该研究分析了各种Si-Fe(II)共沉淀的结构与还原脱卤反应之间的关系。这项工作揭示了Si-Fe(II)相互作用对COCs命运的影响,这对于COCs污染的地下水环境的修复具有重要意义。
图文导读
Si-Fe(II)共沉淀物表征
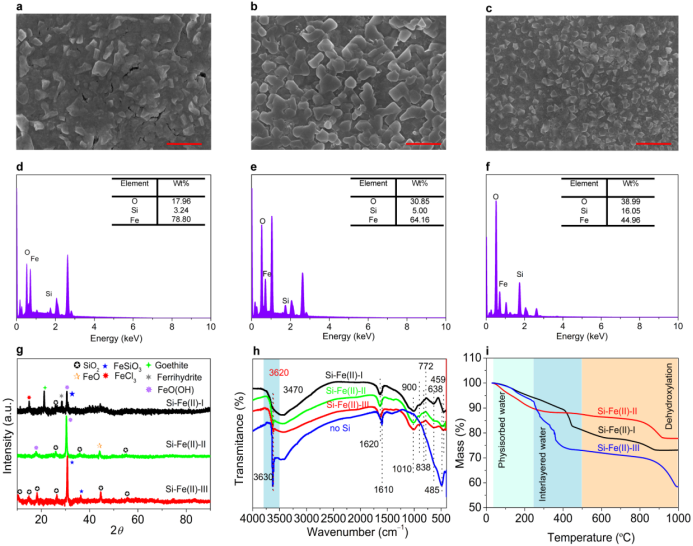
图1. Si-Fe(II)共沉淀物形貌和结构分析. a-f Si-Fe(II)-I, Si-Fe(II)-II 和 Si-Fe(II)-III的SEM-EDS.. g Si-Fe(II)-I, Si-Fe(II)-II and Si-Fe(II)-III的XRD 图. h Si-Fe(II)-I, Si-Fe(II)-II, Si-Fe(II)-III 和 无Si(羟基化亚铁)的红外谱图. i Si-Fe(II)-I, Si-Fe(II)-II 和 Si-Fe(II)-III 的TGA. 实验条件: [Fe(II)]0=20 mM, [Si]0=5 mM, pH=7.
我们通过改变羟基化顺序合成了不同类型的Si-Fe(II)共沉淀物。Si-Fe(II)-III中Si的表面超结构中固有的表面态带更多地位于Si-Fe(II)共沉淀的表面。由于Si的表面超结构的存在,在硅表面形成二维带。带中的电子沿着三维带中的表面态载流子移动。吸附的铁原子还可以将载流子注入表面态带。因此,电导率提高,导致Si-Fe(II)-III的高还原活性。Si-Fe(II)-I和Si-Fe(II)-II为白色沉淀,而Si-Fe(II)-III的颜色在老化20分钟后从白色变为浅灰绿色。Si-Fe(II)-I主要由针铁矿、铁氢化物、氯化铁、FeSiO3、FeO(OH)和结晶不良的SiO2组成。Si-Fe(II)-III的主要成分是FeSiO3和SiO2,其结晶度略高于Si-Fe(II)-II。FTIR和TGA表明,不同羟基化顺序得到的Si-Fe(II)共沉淀物中Fe(II)的配位环境是显著不同的。
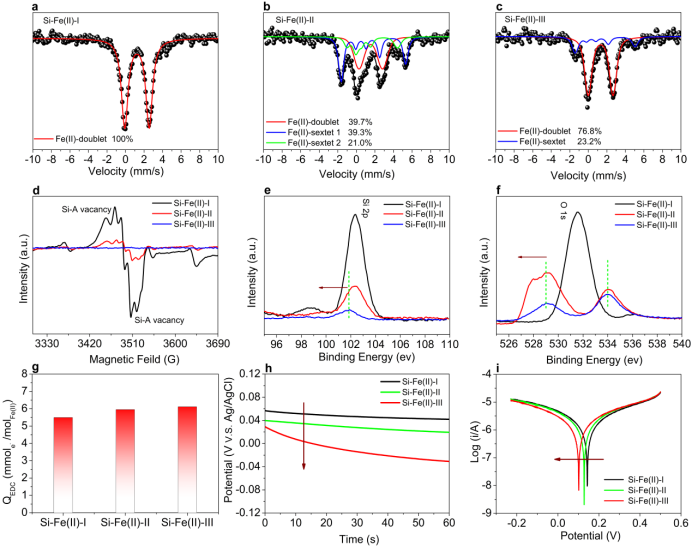
图2. Si-Fe(II)共沉淀物的局域结构、化学态和给电子能力。a-c Si-Fe(II)-I, Si-Fe(II)-II 和Si-Fe(II)-III 在 13 K时的穆斯堡尔谱图. d Si-Fe(II)-I, Si-Fe(II)-II 和Si-Fe(II)-III 在室温时EPR 谱图. e Si-Fe(II)-I, Si-Fe(II)-II 和 Si-Fe(II)-III 中Si 2p 的XPS谱图. f Si-Fe(II)-I, Si-Fe(II)-II 和 Si-Fe(II)-III 中O 1s 的XPS谱图. g ABTS作为介体时的Si-Fe(II)-I、Si-Fe(II)-II和Si-Fe(II)-III的QEDC。 Si-Fe(II)-I、Si-Fe(II)-II和Si-Fe(II)-III在无氧的0.5M Na2SO4 溶液中的h 开路电压 和 i Tafel 曲线. 实验条件: [Fe(II)]0=20 mM, [Si]0=5 mM, pH=7.
在13K下获得了不同羟化顺序的Si-Fe(II)共沉淀物的穆斯堡尔谱(图2a-c)。穆谱结果直接表明在不同羟化顺序的Si-Fe(II)共沉淀物中,Fe(II)周围的局部配位环境明显不同。在Si-Fe(II)-I和Si-Fe(II)-II中发现了与Si-A中心相关的空位,且后者的空位强度较弱。相比之下,Si-Fe(II)-III中没有观察到Si-A空位(图2d)。值得注意的是,与Si-Fe(II)-I和Si-Fe(II)-II相比,Si-Fe(II)-III的O1s和Si 2p XPS峰向较低的结合能移动(图2e-f),这表明在Si-Fe(II)-III中,电子从Fe(II)部分转移到O和Si。这一现象与EPR数据(Si-Fe(II)-III中的Fe(II)具有很强的给电子能力)是一致的。在强氧化剂(ABTS)存在下,Fe(II)在三种不同类型的Si-Fe(II)共沉淀物中的给电子能力是相同的,结构差异对还原反应活性的影响可以忽略。Si-Fe(II)-III的开路电位最低,说明在所研究的Si-Fe(II)共沉淀物中,Si-Fe(II)-III最不稳定。在三种样品,Si-Fe(II)-III的腐蚀电流最高(1.439×10−6A),说明Si-Fe(II)-III具有最高的还原活性。
Si-Fe(II)共沉淀物对CT的降解
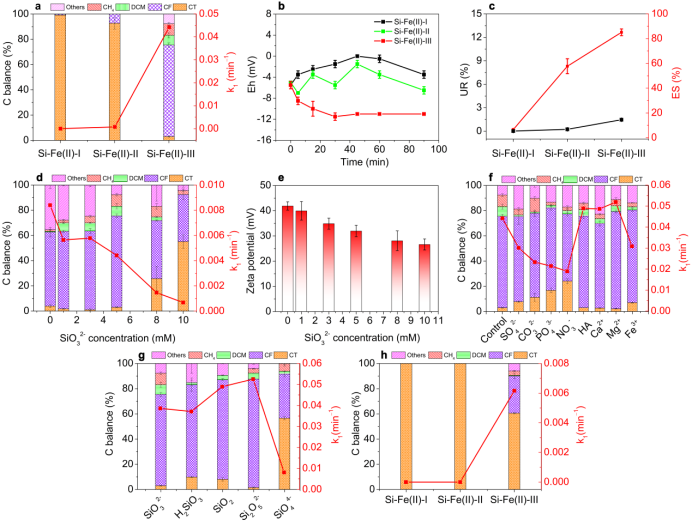
图3. 环境相关条件下Si-Fe(II)共沉淀物还原CT的研究. a 不同Si-Fe(II)共沉淀物对CT还原的产物分布和反应速率, b Eh随时间的变化, c 不同Si-Fe(II)共沉淀物对CT还原的电子利用率和选择性, dSi投加量对脱氯产物分布和CT降解率的影响, e Si投加量对Si-Fe(II)-III共沉淀物Zeta电位的影响([Si]0=0-10 mM), f 离子影响([ions]=1 mM (HA=1 mg/L)), g Si形式影响, h地下水对脱氯产物分布和CT降解率的影响. 实验条件: [Fe(II)]0=20 mM,[CT]0=20 μM, [Si]0=5 mM, 反应时间 = 90 min, pH=7.
Si-Fe(II)-III在90min内将CT还原为CF、DCM和CH4,但Si-Fe(II)-I和Si-Fe(II)-II体系对CT的还原反应活性很弱(图3a)。Si-Fe(II)-I、Si-Fe(II)-II和Si-Fe(II)-III的Eh值差异不显著,说明它们的反应活性差异并不主要是由于Eh值的不同(图3b)。在三种类型的Si-Fe(II)共沉淀物中,Si-Fe(II)-III的利用率(UR,1.47±0.02%)和电子选择性(ES,85.1%)是最高的(图3c)。随着−用量从0.084增加到10 mM,Si-Fe(II)-III还原CT的速率变慢,伪一级速率常数(k1)从0.084 min−1下降到0.00672 min−1 (图3d)。随着Si用量的增加,Si-Fe(II)-III共沉淀物的Zeta电位逐渐降低,且均为正值(图3e)。这表明,随着Si加入量的增加,Si-Fe(II)-III共沉淀物的分散性变差。带正电的Si-Fe(II)表面([FeII-O-Si]+)具有稳定卡宾阴离子的作用。不管这些表面结合中间体的性质如何,这一系列连续的加氢还原最终将形成CH4。无机阴离子SO42−, CO32−, PO43−, NO3−的存在使CT还原速率常数(k1)从0.04419 min−1分别降至0.03013 min−1、0.02331 min−1、0.02145 min−1和0.01896 min−1 (图3f)。而HA、Ca2+、Mg2+和Fe3+的存在则是促进Si-Fe(II)-III共沉淀物对CT的还原脱氯。在Si-Fe(II)共沉淀物/CT体系中,Si的形态对还原反应活性和产物分布有重要影响(图3g)。与超纯水相比,真实地下水中Si-Fe(II)共沉淀物对CT的还原作用受到抑制(图3h)。
机理分析
结构变形、内电场与空位
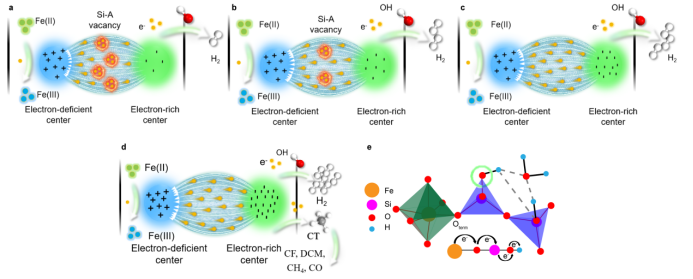
图4. 内电场 a Si-Fe(II)-I, b Si-Fe(II)-III, c Si-Fe(II)-III, d Si-Fe(II)-III 共沉淀物 + CT, e Si-Fe(II)共沉淀物的简化图.
在Si-Fe(II)共沉淀物中,Si-Fe(II)-III中Fe(II)双重态的ΔEQ值较大。对于六配位Fe(II)物种,八面体结构的小范围扭曲导致了较大的四极分裂(ΔEQ)值。因此,Si-Fe(II)-III的Fe(II)八面体的结构变形比Si-Fe(II)-I的小,这可能与Si-Fe(II)-III具有较高的还原活性有关。另一方面,Si-Fe(II)-II和Si-Fe(II)-III分别含有两个和一个Fe(II)六重态。在超精细相互作用中,Fe(II)六重态的中心位移(CS)值越高,Fe(II)的s电子密度就越低。在Si-Fe(II)共沉淀物中,Si-Fe(II)-III的CS值最低,为1.087 mm/s。因此,Si-Fe(II)-III中的Fe(II)可能向O原子提供更多的电子。Fe(II)八面体的结构扭曲可能是由Jahn-Teller效应和空位引起的。结果表明,空位浓度按Si-Fe(II)-I>Si-Fe(II)-II>Si-Fe(II)-III的顺序递减,这与结构扭曲程度的大小顺序相同。我们的结果与前人的研究结果之间的矛盾表明,决定Si-Fe(II)共沉淀物结构扭曲的是Jahn-Teller效应,而不是空位。Si-Fe(II)-III的低结构扭曲程度可能导致其较高的还原反应活性。
Si-Fe(II)共沉淀物表面Fe物种在724.8-723.4 eV和709.9-711.3 eV的结合能可分别分配给Fe(II)的2p3/2和2p1/2(图S3a)。这一结合能高于FeO中Fe(II)的结合能(722.8和709.8 eV),证实了Si-Fe(II)共沉淀物中Fe位附近的缺电子性质。因此,在Si-Fe(II)共沉淀结构中,较高的电子密度集中在与晶格Si相邻的晶格O2−中。较低的电子密度分布在Fe位置附近,形成无数的内电场。富电子的O中心充当阴极,晶格Fe充当阳极。内电场的方向是从富电子中心到贫电子中心(图4a-d)。缺电子的Si-A空位容易在Si-Fe(II)结构中提取/捕获电子,抑制了可能的电子从Fe(II)到与Si原子相邻的富电子晶格O2−的转移。因此,电子密度最高的区域离Si更近。四面体Si周围电荷密度的增加自发地增加了其邻近O原子的电子密度。这导致连接到O原子的H原子首先得到电子(图4e),连接到四面体Si的羟基被还原为H2。因此,Si-Fe(II)-III的最高反应活性源于其独特的电子结构(图2)。
表面羟基和吸附CT增强内电场
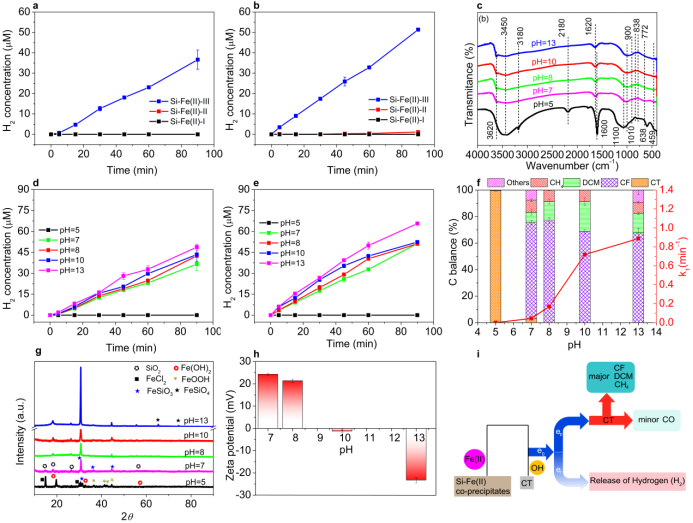
图5. 表面羟基和吸附态CT强化内电场. a不同Si-Fe(II)共沉淀物的H2释放 ([Fe(II)]0=20 mM, [Si]0=5 mM, 反应时间 = 90 min, pH=7). b CT存在时不同Si-Fe(II)共沉淀物的H2释放([Fe(II)]0=20 mM, [CT]0=20 μM, [Si]0=5 mM, 反应时间= 90 min, pH=7). c Si-Fe(II)-III 共沉淀物在不同pH下的FTIR 光谱. d 不同pH条件下Si-Fe(II)共沉淀物的H2释放([Fe(II)]0=20 mM, [Si]0=5 mM, 反应时间 = 90 min, pH=5, 7, 8, 10 and 13). e CT存在时,不同pH条件下Si-Fe(II)共沉淀物的H2释放([Fe(II)]0=20 mM, [CT]0=20 μM, [Si]0=5 mM, 反应时间 = 90 min, pH=5, 7, 8, 10 and 13). f不同pH值下Si-Fe(II)-III还原脱氯反应的产物分布和反应速率. g 不同pH值下Si-Fe(II)-III的XRD. h 不同pH值下Si-Fe(II)-III的zeta电位. i不同Si-Fe(II)共沉淀物的电子平衡. 实验条件: [Fe(II)]0=20 mM, [Si]0=5 mM, 反应时间= 90 min, pH=5, 7, 8, 10, 13.
Si-Fe(II)共沉淀物本身可以通过表面羟基的氧化产生氢气(图4a-c和图5a)。随着时间的延长,H2的释放浓度逐渐增加。此外,与Si-Fe(II)-I和Si-Fe(II)-II相比,Si-Fe(II)-III在90min时释放的H2相对较高。这是因为Si-Fe(II)-III不仅表面羟基含量最高(图1i),而且还原强度最强(图4c)。CT的加入促进了体系中氢气的产生(图5b)。CT的氯是一个亲电基团,这导致富电子区域的电子密度增加,表面羟基被氧化。此外,随着pH的升高,Si-Fe(II)-III释放的H2逐渐增加(图5d)。在相同的pH条件下,CT的存在促进了H2的产生(图5e)。这一现象进一步证明了羟基浓度的增加和CT的存在可以增强内电场。Si-Fe(II)-III共沉淀物在pH值为5时完全溶解,导致还原反应活性降低(图5f)。在酸性条件下,Si-Fe(II)不与表面羟基配位,不形成内部电场。另一方面,随着pH的升高,Si-Fe(II)-Ⅲ对CT的还原速率逐渐增加(图5f和S18),并在pH为13(k1=0.88738 min−1)时达到最大。随着pH值的升高,Si-Fe(II)-III主要为FeSiO3,结晶度增加,少量结晶不良的SiO2(图5g)。这一现象证实了FeSiO3在Si-Fe(II)-III还原CT过程中的重要作用。随着pH的升高,Si-Fe(II)-III的Zeta电位逐渐由正向负转变(图5h)。此外,Fe(II)(e0)给出的电子数是CT还原(ep)和氢释放反应(ei)消耗的电子数之和(图5i)。这进一步揭示了表面羟基和CT消耗相同的Fe(II)电子源进行进一步的转化。
小结
这项研究证明了在可能的自然地下环境中形成的Si-Fe(II)共沉淀物的结构和反应活性的多样性。不同的硅羟化顺序对氧化还原活性Fe(II)中心的配位环境有显著影响。不同的合成方法制备的Si-Fe(II)共沉淀物的物相组成有显著差异。Si-Fe(II)共沉淀物由无数的内电场组成,羟基的转化和四氯化碳(CT)的转化都消耗了电子。然而,羟基和CT的共存由于电负性增加了富电子区域的电子密度,增强了它们的接受电子的能力。与其他化学成分相同的Si-Fe(II)共沉淀物相比,Si-Fe(II)-III的结构扭曲程度低,空位浓度低,表面羟基和吸附的CT分子增强了内电场,使其具有较高的还原反应活性。
本项目得到了国家自然科学基金委、中央高校基本科研业务费专项资金项目和js6666zs金沙人才引进启动经费。
通讯作者简介

黄理志:js6666zs金沙研究员、博导,湖北省高层次人才,中国水协青年委委员,国际水协会员,主持多项国家级科研项目,发表论文50余篇,以第一/通讯作者在Environmental Science & Technology、Water Research等期刊发表SCI论文41篇(其中35篇中科院一区、33篇影响因子>10、1篇ESI高被引);授权发明专利4项,技术应用2项,成果转化1项。指导学生科研竞赛获国家级奖励7项,省部级奖励6项。
课题组网站:https://www.x-mol.com/groups/lizhihuang
第一作者:贾欠欠,女,博士研究生,现就读于js6666zs金沙。
文章链接:https://doi.org/10.1016/j.watres.2022.119386







